A high-quality genome assembly and its detailed annotation are keys in supporting the development of genomic tools for applications in spruce breeding programs, and for improving our fundamental understanding of conifer genome structure and functions.
In previous work in the SMarTForests Project (www.smartforests.ca), we developed genome assemblies for white spruce and interior spruce genotypes used in Canadian spruce breeding programs. These and other conifer genome assemblies represent different levels of completeness. In general, improvements in assembly contiguity of conifer genomes are much needed, including improved contiguity of the genic space. This activity of the Spruce-Up Project builds on the fact that improvements in sequencing technologies and new algorithm paradigms offer opportunities to substantially improve such assemblies.
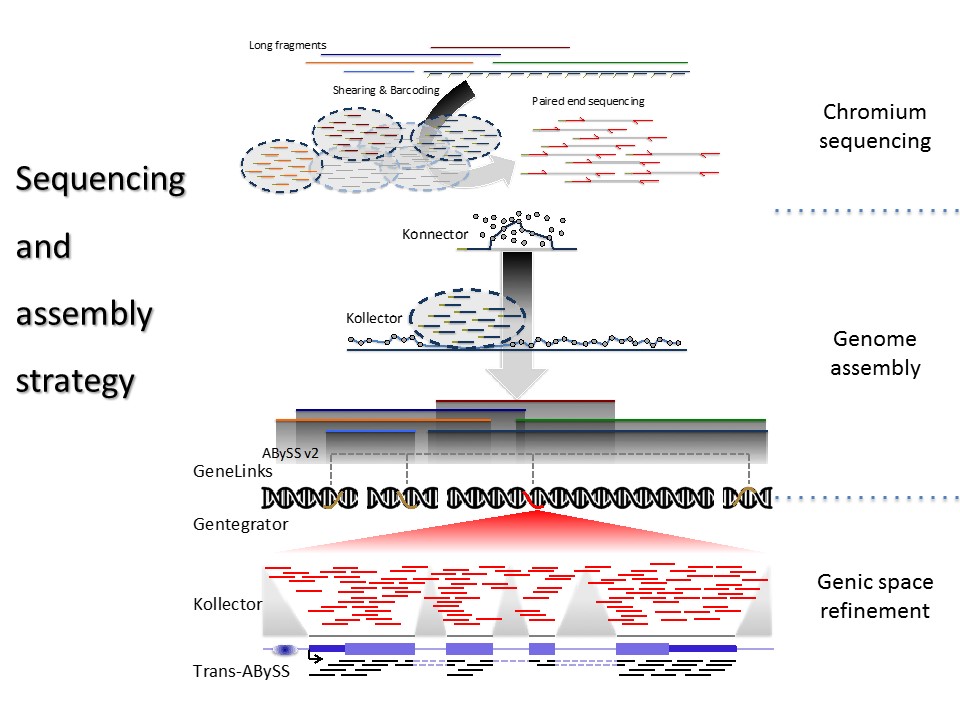
Using established (Illumina) and new sequencing platforms (10x and Oxford Nanopore), we are generating data to bridge gaps in the existing and new spruce genome sequence assemblies for white spruce, interior spruce and Sitka spruce.
We are developing algorithms for long pseudo-read manipulation (Konnector) and focus on scaffold gap closing (Sealer) using our existing bioinformatics programs. We are developing scalable algorithms for read alignments to large non-static targets (DIDA). We are also developing a gene identifier framework that will be stable across assemblies, alleviating challenges around annotation lift-overs between assembly versions (UniqTag).
Objectives
- Deliver high-quality genome sequence assemblies for white spruce, interior spruce, and Sitka spruce.
- Develop detailed annotations for genes and SNPs, and develop a new Sitka spruce genotyping tool, targeting gene SNPs with our partner End-Users and international collaborators.